



PCR是聚合酶链式反应(Polymerase Chain Reaction)的简称,是一种体外扩增特定DNA片段的分子生物学技术,其原理类似 DNA 分子的天然复制过程,是将在待扩增的DNA片段和与其两侧互补的两个寡聚核苷酸引物,经变性、退火和延伸若干个循环后,DNA扩增2n 倍。PCR技术自问世以来,在生物科学领域、分子诊断领域、亲子鉴定、法医鉴定以及犯罪调查等方面发挥了巨大作用,是迄今为止最为重要的技术之一。经过演化和革新,PCR技术已经发展了三代:普通PCR技术、实时荧光定量PCR技术(qPCR)以及数字PCR(dPCR)技术。
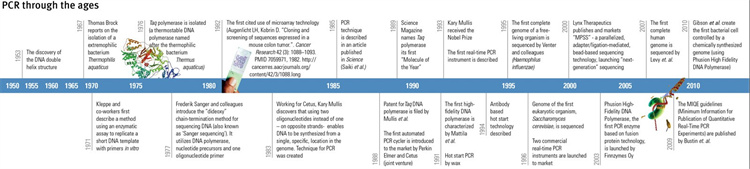
图1 PCR 的演变(1950–2010年)
PCR的基本原理与DNA的体内复制相类似,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性、退火和延伸三个基本反应步骤构成:
①模板DNA的变性:模板DNA经高温(95℃左右)加热一定时间后,使其解离成单链,以便它与引物结合;
②模板DNA与引物的退火(复性):将温度降至55℃左右时,引物与模板DNA单链按碱基互补配对的原则结合;
③引物的延伸:再将温度调至72℃左右(DNA聚合酶最适反应温度),DNA模板和引物结合物在Taq DNA聚合酶的作用下,按碱基配对与半保留复制原理,沿着磷酸到五碳糖(5'→3')的方向合成一条新的与模板DNA链互补的半保留复制链。这样经变性、退火和延伸重复若干个循环后,就能将待扩目的基因扩增放大几百万倍。
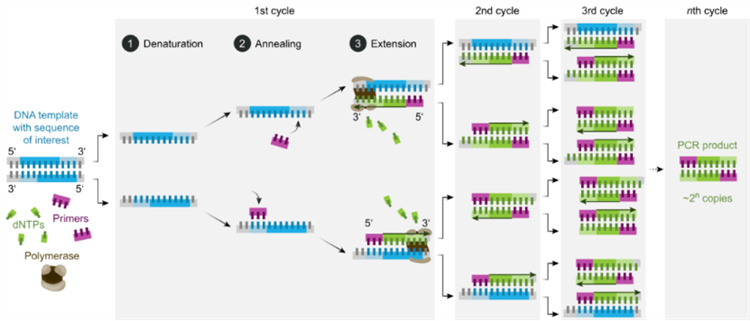
图2 PCR的基本反应步骤
1.普通 PCR
普通PCR即一代PCR,使用普通PCR扩增仪扩增靶基因,并通过琼脂糖凝胶电泳对产物进行定性分析。
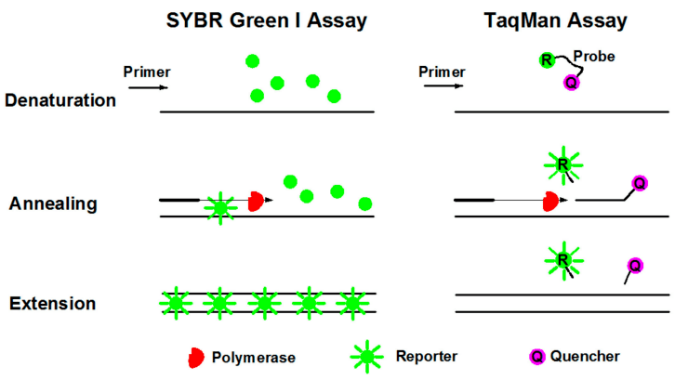
2.实时荧光定量PCR(qPCR)
实时荧光定量PCR,也叫Real-Time PCR,即二代PCR,是指在PCR扩增反应体系中加入荧光染料或者荧光基团,在整个PCR过程中通过收集荧光信号实时监测每一个循环中扩增产物量的变化,最后通过标准曲线和CT值对待测样品进行定量分析。qPCR常用的有两种方法:SYBR Green法和TaqMan探针法。①荧光染料法(SYBR Green):SYBR Green Ⅰ是荧光定量PCR中最常用的荧光染料,它能与所有的双链DNA结合。在PCR反应体系中,加入SYBR Green Ⅰ,它就会在过程中与双链DNA结合,从而产生荧光信号。因此,反应中发出的全部荧光信号就会与反应中双链DNA的量呈正比,荧光强度也会随着产物的增加而增加。但是由于染料与双链DNA是非特异性结合,因此可能产生假阳性的结果。
②荧光探针法(TaqMan技术):TaqMan探针是最早用于定量的方法,也是临床检测中最常用的检测方法。PCR扩增时,加入一对引物的同时再加入一个特异性的荧光探针。该探针是一个寡核苷酸,5'端标记一个荧光报告基团(Reporter, R),3'端标记一个淬灭基团(Quencher, Q)。当探针完整时,报告基团发射的荧光信号被淬灭基团吸收,以至于无法检测到荧光信号;而当PCR扩增时(在延伸阶段),探针会被Taq酶的5'→3'外切酶活性酶切降解,使报告基团和淬灭基团分离,报告基团发射底的荧光不会再被吸收,从而可以在荧光监测系统接收到荧光信号,即每扩增一条DNA,就形成一个荧光分子,PCR产物的形成与荧光分子的形成完全同步,PCR产物越多,荧光信号累积的越多,荧光强度越大。
注:多重PCR,也称多重引物PCR或复合PCR,是在一个反应体系中加入两对以上引物,同时扩增出多个核酸片段的一种新型PCR扩增技术。
3.数字PCR(Digital PCR, Dig-PCR, dPCR)
数字PCR即三代PCR,是对原始PCR的另一种改进,是一种能够实现核酸绝对定量的精准检测技术,基于泊松分布原理将核酸样品分配到大量独立、平行的微反应单元(纳升级)中,使每个反应室中平均只有一个拷贝或者没有目标DNA分子,然后加入荧光信号进行扩增,从而实现靶标核酸分子的绝对计数,提高检测灵敏度和准确度。
4. 逆转录PCR( RT - PCR)
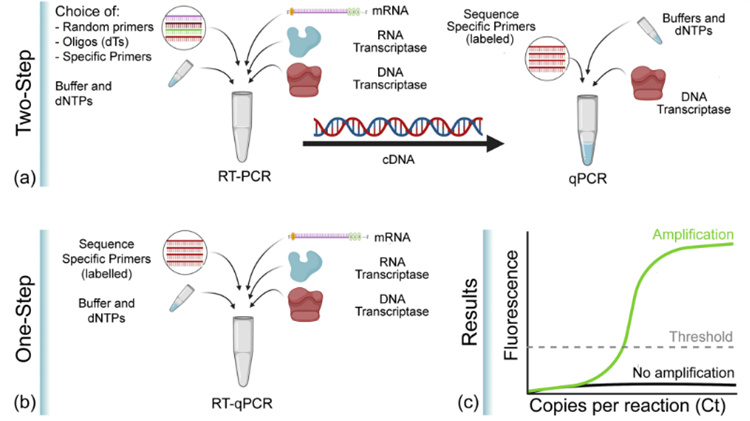
逆转录PCR也叫反转录PCR,将RNA的反转录(RT)和cDNA的聚合酶链式扩增相结合,是PCR的一种广泛应用的变形。在RT-PCR中首先经反转录酶将一条单链RNA逆转录成cDNA,再以cDNA为模板,扩增合成目的片段。作为模板的RNA可以是总RNA、mRNA或体外转录的RNA产物。无论使用何种RNA,关键是确保RNA中无RNA酶和基因组DNA的污染。RT-PCR技术灵敏且应用广泛,可用于检测细胞中基因表达水平,细胞中RNA病毒的含量和直接克隆特定基因的cDNA序列。一般通过一步法或两步法进行,一步法是RT反应和PCR反应在同一试管中进行;而在两步法中两个反应是分开依次进行的。
5.实时荧光定量逆转录PCR(RT-qPCR)
Real-time RT-PCR是qPCR和RT-PCR的组合,其中的“RT”是Reverse transcription(逆转录)的意思,所以RT-qPCR就是结合了荧光定量技术的逆转录PCR,即以mRNA或总RNA为模板,先反转录得到cDNA,再以cDNA为模板,通过荧光定量PCR进行定量检测分析。因为RT-PCR只可以定性,但不能进行定量分析。与RT-PCR一样,RT-qPCR定量分析RNA也有两种方法:一步法和两步法。两种方法都需要先将RNA反转录为cDNA,然后再将其作为qPCR扩增的模板,只是一步法中的RT和qPCR在同一试管中进行,两步法中的RT和qPCR是按顺序分开进行。
凯斯艾生物依托先进的技术平台和丰富的项目经验,致力于为客户提供高灵敏度、高特异性的实验方案,助力基因表达分析、病原体检测、突变筛查等多个方向的科学研究与临床转化,具备如下检测能力:
• 引物设计和验证
• 核酸提取(对于非致病微生物、病毒等):试剂盒法、Trizol法
• 琼脂糖凝胶电泳:RNA、DNA电泳
• 核酸浓度检测
• RT-PCR:猫FIPV筛选
• 基因表达分析:
相对定量法,如组织、BALF细胞、培养的细胞等
绝对定量法,如病毒滴度、组织分布等
• qPCR方法开发和验证
• 其他,如支原体检测(化学发光法、qPCR法)
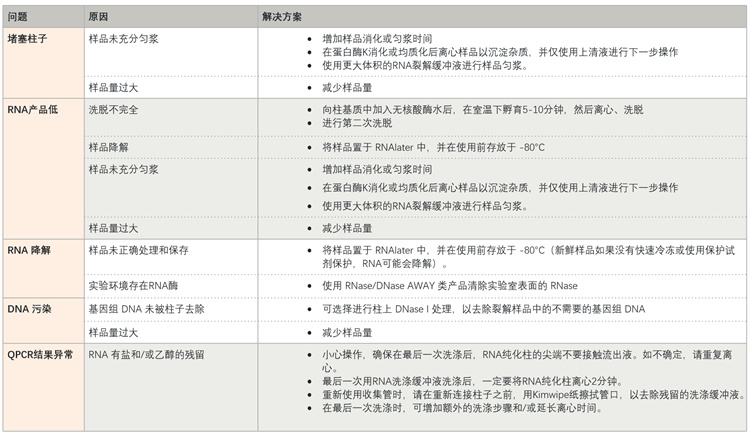
1.RNA提取常见问题
2.怎么判定RNA质量?
2.1 Nanodrop 检测吸光度
Table 1. 核酸理想吸光度比值范围
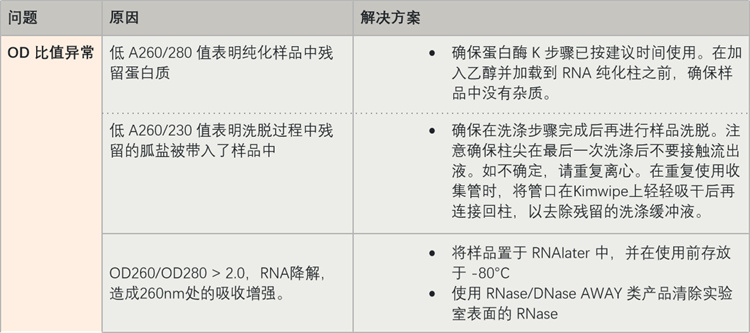
Table 2. 异常吸光度比值可能原因
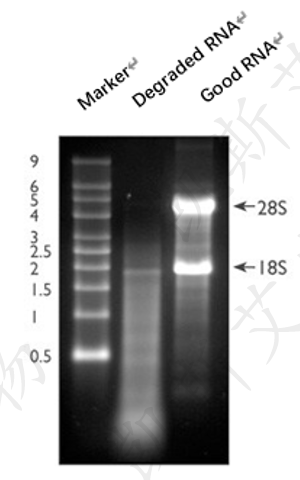
评估总RNA完整性的最常用的方法是使用少量的RNA样品进行变性凝胶电泳并用溴化乙锭(EtBr)进行染色。完整的总RNA在进行变性凝胶电泳时会产生清晰的28S和18S rRNA条带(真核样品)。28S rRNA条带的强度应当大致为18S rRNA条带的两倍(图5,第3条泳道)。这种2:1的比率(28S:18S)是判断RNA完整性的一个很好的指标。部分降解的RNA样品电泳条带会弥散,没有清晰的rRNA条带,或者不会出现前面提到的高质量RNA才具有的2:1比率(28S:18S)。完全降解的RNA会表现为在极低分子量处的弥散状(图5,第2条泳道)。
3.QPCT 结果显示样品Ct值不稳定
Ct值飘忽不定,各种污染反反复复,做qPCR实验操作时要特别注意三个部分--防污染、混匀操作和实验细节的处理。
3.1 防污染为什么如此重要呢?因为qPCR是一种灵敏度非常高的实验技术,一旦有污染出现之后进行去除是非常困难非常麻烦的,所以一定要注意防污染操作。
• 实验室应尽量进行严格的分区,包括RNA提取的区域,反应液配置区,模板添加区。那么在反应液配置区,一定不要有核酸的出现,RNA的提取和模板添加的操作最好在生物安全柜中进行。相应的设备、器具、试剂和耗材等都要分区来使用。
• 在进行加样的时候,枪头盒必须要保持关闭的状态,也不可以在吸完阳性对照之后经过打开的枪头盒上方,这些都是可能会引起污染的。
• 在加样顺序方面,应在反应液配置区添加阴性对照并盖好盖子后,再拿到模板添加区加入待检核酸,最后再加阳性对照。
3.2 在试剂的混匀操作使用方面需要注意。我们使用的试剂大部分都是预混型的,为了保护试剂中的酶通常还会加入甘油等粘度较大的物质,试剂使用前一定要在冰上完全融化并且混匀后再使用,有的老师因为没有将试剂混匀而导致实验结果不佳。混匀时要注意轻柔的混匀,不要使用振荡器,因为试剂中含有酶,使用振荡器剧烈混匀的话可能会导致酶活下降。
• 首先是体积不超过100 μl的组分,可以用手轻弹管底,使液面上下浮动,要尽量避免起泡,再使用桌面小离心机进行瞬时离心。
• 体积较多的超过100μl的组分,采用边颠倒边轻弹的方式混匀,也要注意尽量避免产生气泡,颠倒10次左右,再使用桌面小离心机瞬时离心。
• 在进行模板稀释的时候也应该采用轻弹离心的方式进行混匀。
3.3 最后一个需要注意的方面,是要在实验操作中注意实验细节的处理。保持良好的操作习惯,这样可以减少平行样之间的操作误差。
• 可以通过先配置预混液分装再加入模板的方式减少误差。配置预混液时需要先加入体积最大且最稳定的组分——通常是水,而体积最小且最不稳定的组分需要最后加入——常常是酶,预混液也要记得进行颠倒混匀或者使用移液器吸打混匀之后再进行分装。• 一些试剂或者模板/引物的加入量很少,移液器是否准确也是一个关键问题,一个很小的误差经过指数放大之后就会变得明显,对Ct值的影响还是非常大的,而且平行样之间如果差距比较大,和移液器是否正确使用、有没有定期校正都是有关系的。